| Description | Unlike most treatments of insomnia that target the GABA (g-aminobutyric acid) receptor complex, ramelteon is an agonist of the melatonin receptor. In particular, it has high selectivity for the MT1 and MT2 subtypes, which have been implicated in the maintenance of circadian rhythms, over the MT3 receptor responsible for other melatonin functions. Its lack of affinity for not only the GABA receptor complex but also neurotransmitter, dopaminerigic, opiate, and benzodiazepine receptors suggests an improved safety profile devoid of the abuse potential of the hypnotic drugs that target these receptors. As such, ramelteon is not a scheduled drug. Primary metabolites include hydroxylation and oxidation to carbonyl species with secondary metabolites resulting from glucuronidation. Since CYP1A2 is the major isozyme involved in the hepatic metabolism of ramelteon, it should not be taken in combination with strong CYP1A2 inhibitors, such as fluvoxamine. Co-administration with either ketoconazole (a CYP3A4 inhibitor) or fluconazole (a potent CYP2C9 inhibitor) resulted in significant increases in AUC and Cmax, but the extensive metabolism and highly variable plasma concentrations of ramelteon precluded the need for dose modification. The package insert, however, cautions patients about co-administration with potent CYP3A4 and CYP2C9 inhibitors. Based on the result of the clinical trials, the recommended dose of ramelteon is 8mg taken within 30 min of going to bed. In addition to the precaution of co-administration with CYP inhibitors, it should not be used in patients with severe hepatic impairment. The adverse events, observed in 5% of patients in clinical studies, were somnolence, dizziness, nausea, fatigue, headache, and insomnia. |
| Chemical Properties | Crystalline Solid |
| Originator | Takeda (Japan) |
| Uses | Melatonin MT1/MT2 receptor agonist. Sedative, hypnotic |
| Uses | Melatonin MT1/MT2 receptor agonist. Sedative, hypnotic. |
| Uses | melatonin receptor agonist |
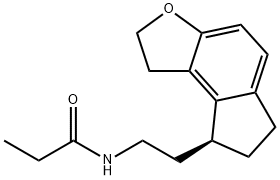
| Definition | ChEBI: N-[2-[(8S)-2,6,7,8-tetrahydro-1H-cyclopenta[e]benzofuran-8-yl]ethyl]propanamide is a member of indanes. |
| Brand name | Rozerem (Takeda). |
| General Description | The melatonin molecule wasmodified mainly by replacing the nitrogen of the indole ringwith a carbon to give an indane ring and by incorporating 5-methoxyl group in the indole ring into a more rigid furan ring.The selectivity of the resulting ramelteon for MT1 receptor iseight times more than that of MT2 receptor. Unlike melatonin,it is more effective in initiating sleep (MT1 activity)rather than to readjust the circadian rhythm (MT2 activity). Itappears to be distinctly more efficacious than melatonin butless efficacious than benzodiazepines as a hypnotic.Importantly, this drug has no addiction liability (it is not acontrolled substance). As a result, it has recently been approvedfor the treatment of insomnia. |
| Synthesis | Vilsmeier-Haack reaction on benzofuran 112 provided aldehyde 113 (100%), which was converted to olefin 114 (88%) by Horner-Emmons reaction with triethylphosphonoacetate, and was followed by hydrogenation of the olefin to give ester 115 (100%). In order to avoid the cyclization of the acid chloride intermediate into the wrong position, the benzene ring was protected by bromination. Both bromination and hydrolysis of the ester is accomplished in a single pot to give acid 116. Thus the ester is brominated with bromine in sodium acetate and acetic acid at 0??C and RT for several hours followed by quenching of remaining bromide by sodium thiosulfate. The resulting acidic solution was taken up in acetonitrile and refluxed for 2hr to provide the acid 116 in 73% yield. The conversion of the acid to acid chloride was done by reacting with thionyl chloride in odichlorobenzene at 40??C for 30 to 40 min after which the reaction was cooled to 0??C . Aluminum trichloride was added and the reaction mixture was stirred at 0??C for 30 min to deliver cyclized ketone 117 in 92% yield. After completion of the cyclization, the bromines are removed by hydrogenation (86%) and resulting ketone 118 was then reacted under Horner-Emmons condition with diethyl cyano phosphonate to give vinyl nitrile 119 in 84% yield. Selective reduction of the nitrile was accomplished by hydrogenation under basic condition (sodium hydroxide in toluene) in the presence of the activated cobalt at 25-50??C for 6.5 hr. The amine was recovered as hydrochloride salt 120 (99% yield) by treating the amine with HCl in methanol. In the next step, the amine salt 120 was taken up in toluene and treated with sodium hydroxide followed by hydrogenation of the mixture with [RuCl(benzene)(R)-BINAP]Cl as catalyst to provide chiral amine 121, after several work up and palladium catalyzed hydrogenations, in 73% overall yield. Final acylation of the amine with propionyl chloride in the presence of aqueous sodium hydroxide in THF at room temperature gave the desired product ramelteon (XVI), after crystallization, in 97% yield.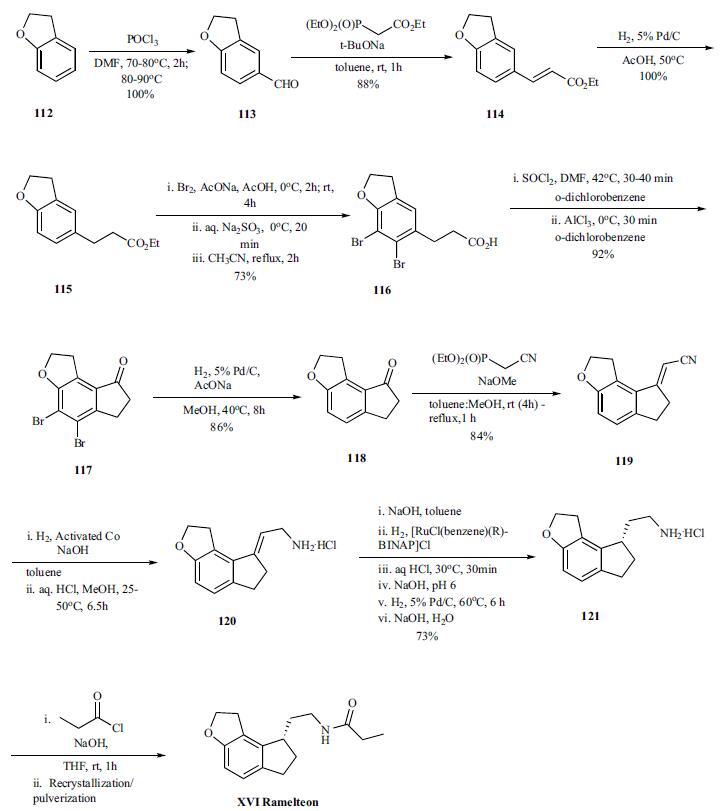 |
| references | [1] miyamoto m. pharmacology of ramelteon, a selective mt1/mt2 receptor agonist: a novel therapeutic drug for sleep disorders. cns neurosci ther. 2009 winter;15(1):32-51. doi: 10.1111/j.1755-5949.2008.00066.x. |